The Big Data Department of CNIC has gained new progress in supporting the construction of the microbiological field database and its analysis system
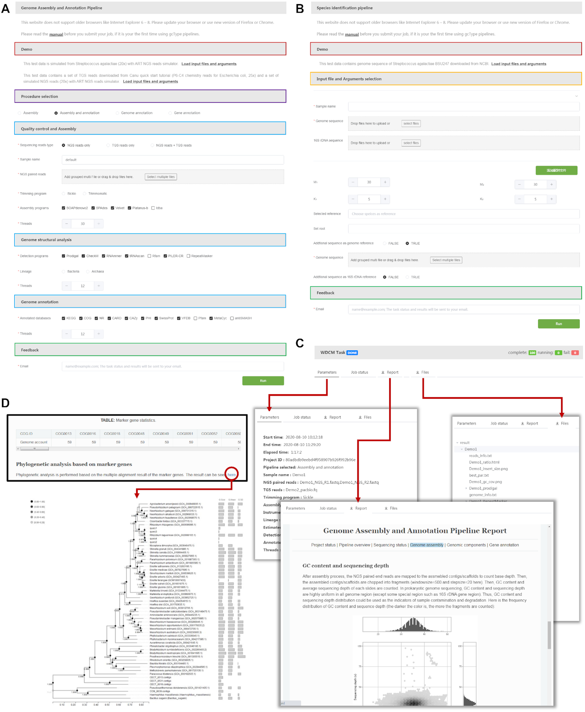
Submission pages and results of the analysis for the two pipelines, provided by gcType
The Big Data Department of CNIC and the team of researcher Ma Juncai under the Institute of Microbiology, CAS have built the Global Catalogue of Type Strain (gcType), which integrates more than 13,944 genome data of 16701 effectively published prokaryotes. gcType is currently the most comprehensive and functional data platform with regard to the model Microbial genome data that provides users with one-stop data management, genome annotation, and new species identification analysis. The results of the cooperation are published in the internationally renowned academic journal Nucleic Acids Research.
As the global new coronavirus genome continues to spread, the new coronavirus genome continues to mutate during the epidemic. In addition to the collection and display of data, these databases contain functions such as virus typing and traceability analysis, therefore providing important information for the monitoring and tracking of the global epidemic. However, with the in-depth study of mutation, the functional impact of mutation has gradually become the focus of attention. At present, multiple infectivity-enhancing mutation axes, including Alpha, Beta, and Delta poisoning, have been discovered in many countries and regions around the world. The risk of immune escape may reduce the protection of disease control methods, affect the applicability of disease diagnosis, and evacuate the epidemic. Therefore, the existing database that focuses on data collection and display cannot meet the needs of the future. A virus mutation assessment and systematic early warning system based on big data is needed to systematically evaluate and interpret the impact of various mutations that may occur in the present and in the future, so as to form an effective epidemic prevention and control strategy.

Features of the variations evaluation and prewarning system (VarEPS) portal
The Big Data Department of CNIC, with cooperation of researcher Ma Juncai under the Institute of Microbiology, CAS and other teams have released the "New Coronavirus Variation Evaluation and Early Warning System" (SARS-CoV-2 Variation Evaluation and Prewarning System), referred to as the VarEPS database. VarEPS is the world's first system for multi-dimensional risk assessment and early warning of known and virtual variants in the SARS-CoV-2 genome. Starting from the perspectives of genomics and structural biology, VarEPS conducts multi-dimensional evaluation of mutations based on the evaluation of the frequency of mutation sites including the difficulty of nucleotide mutations, the difficulty of amino acid substitution, the effect of mutations on the secondary structure of proteins, and the effects of single amino acid mutations , so as to comprehensively analyze the effects of known mutations and potential virtual mutations on the function of the virus. On this basis, the system uses an artificial intelligence classifier algorithm to effectively group mutant strains in terms of spreadability and affinity for neutralizing antibodies, and realizes risk assessment and early warning based on virus sequences. The results of the cooperation were published in the internationally renowned academic journal Nucleic Acids Research.
For details, please contact Ms. Meng Zhen (zhenm99@cnic.cn).